
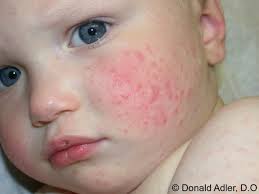

A síndrome de Gianotti-Crosti foi descrita
pela primeira vez em 1955 por Gianotti como uma erupção
papulosa, monomorfa, autolimitada, simetricamente distribuída
na face, região glútea e nas extremidades, ocorrendo
em crianças de dois a seis anos de idade. Em 1970, Gianotti
e Crosti associaram-na à infecção pelo vírus da hepatite B.
Posteriormente foram observadas erupções cutâneas idênticas a Sindrome de Gianotti-Crosti, mas sem evidência de infecção aguda pelo
vírus da hepatite B. Durante a investigação etiológica dos
casos verificamos que são decorrentes
de diferentes agentes infecciosos, principalmente virais .


Na literatura , há apenas duas referências ao HHV6 ( Hepes Virus 6 Humano )como causador da doença.
O curso da Sindrome é benigno e autolimitado, desaparecendo
as manifestações em prazo que varia de duas a oito
semanas, sem recorrências. As lesões cutâneas são
em geral assintomáticas, excepcionalmente pruriginosas. Por vezes há pródromos sugestivos de infecção respiratória
alta. O estado geral mantém-se inalterado ou ocorrem
sintomas tais como mal-estar, febre, náuseas, vômitos,
hepatoesplenomegalia, linfadenopatia e/ou quadro de hepatite
anictérica aguda. Uma linfocitose, eventualmente com
presença de linfócitos atípicos no sangue periférico, pode
ainda ser observada.



 O diagnóstico diferencial deve ser feito com prurigo
agudo, escabiose e outras ectoparasitoses, granuloma anular,
dermatite atópica, líquen plano, erupção liquenóide a
droga, líquen estriado, líquen nítido, histiocitose de células
de Langerhans, pitiríase liquenóide e varioliforme aguda
(Pleva), pitiríase rósea, urticária papular, eritema multiforme
e púrpura de Henoch-Schönlein, entre outros.
O diagnóstico diferencial deve ser feito com prurigo
agudo, escabiose e outras ectoparasitoses, granuloma anular,
dermatite atópica, líquen plano, erupção liquenóide a
droga, líquen estriado, líquen nítido, histiocitose de células
de Langerhans, pitiríase liquenóide e varioliforme aguda
(Pleva), pitiríase rósea, urticária papular, eritema multiforme
e púrpura de Henoch-Schönlein, entre outros.












